Case of the Week #547
Consultant in Maternal-Fetal Medicine, Liverpool Women's Hospital, UK
Posting Dates: October 15 - October 31, 2021
A 25-year-old, primiparous, healthy Caucasian woman in a non-consanguineous relationship was referred to our fetal medicine unit at 22 weeks of pregnancy following concerns with the fetal anatomy scan performed at 20 weeks gestation. The patient had a low risk 1st trimester screen. The key ultrasound findings and fetal biometry are shown below.

View the Answer Hide the Answer
Answer
We present a case of Schinzel Gideon Syndrome
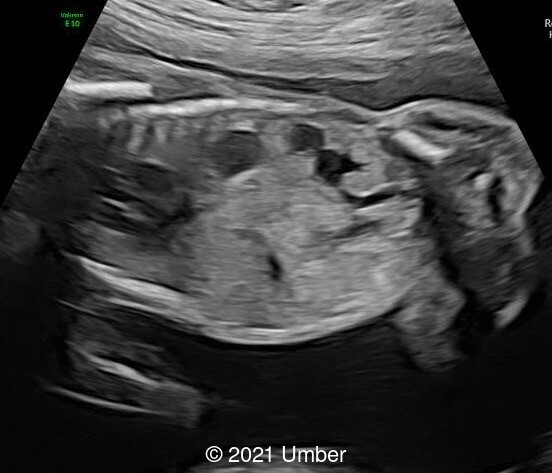
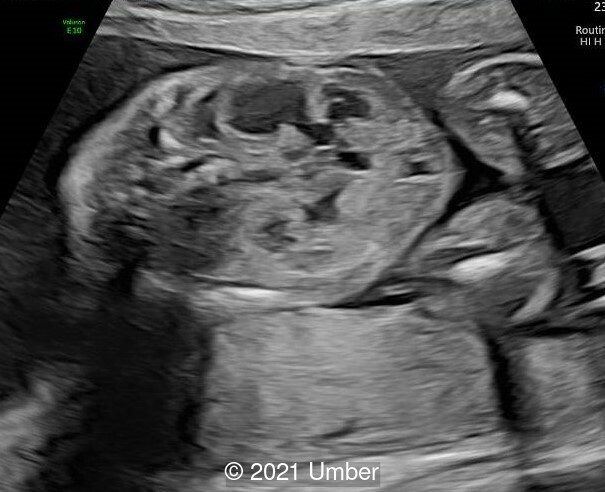
The ultrasound showed the following:
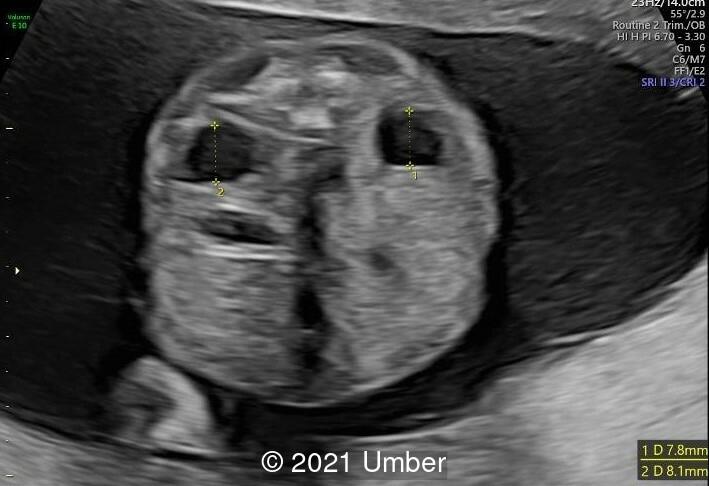
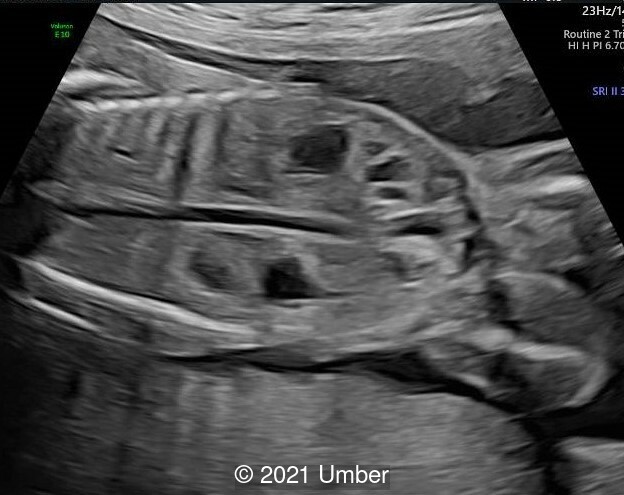
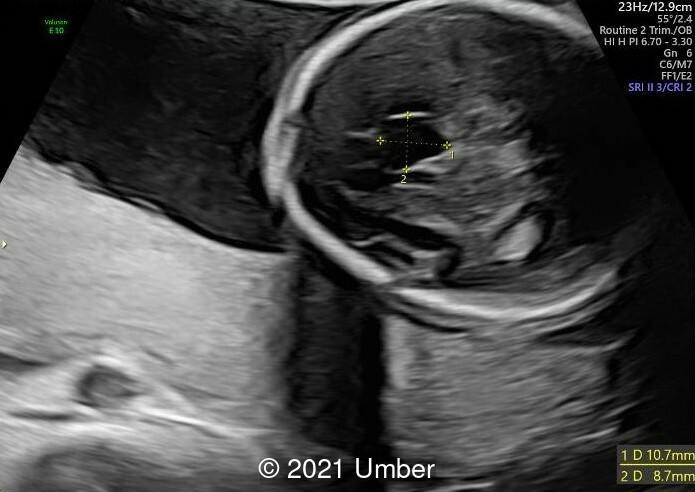
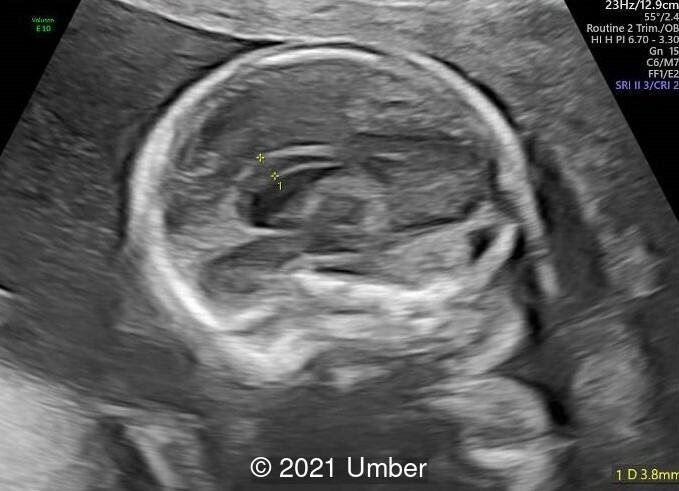
- Bilateral duplex kidney with dilated upper renal pelvises (7.8 and 8.1 mm) and ureters. It appeared that each side had a single ureter that drained both portions of the kidney.
- The bladder was normal.
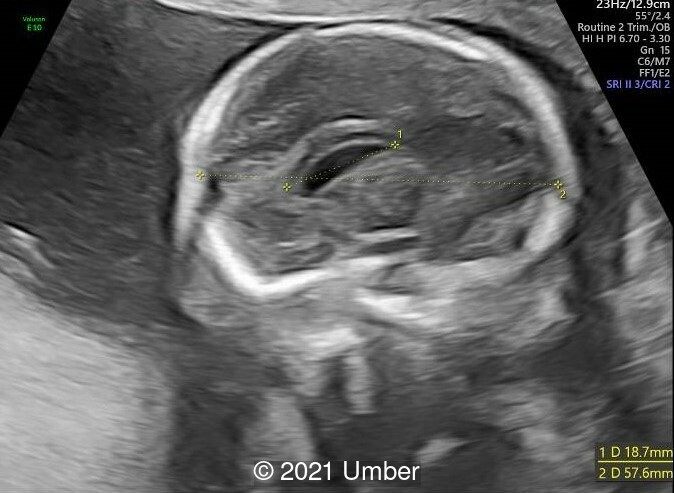
- Cavum septi pellucidi appeared dilated (10x8mm) with slight asymmetry of anterior complex.
- The corpus callosum was seen in its entire length and appeared slightly thickened in middle portion.
- Head circumference was found to be <3rd percentile.
- The rest of the fetal anatomy appeared normal.
After counselling the parents, amniocentesis was performed. Karyotype and microarray were normal. Fetal brain MRI showed biometry of the head circumference to be at lower limit of normal, prominent cerebrospinal fluid spaces, lack of sylvan fissure development, and suspicion of cortical malformation, possibly polymicrogyria. There was concern for hypogenesis of the genu of the corpus callosum. Whole exome sequencing showed a pathogenic variant identified in SETBP1, which is an autosomal dominant, de novo heterozygous mutation associated with Schinzel-Giedion syndrome. After counselling by fetal medicine and genetics, parents opted for termination of pregnancy. As a confirmatory genetic diagnosis was already done, an autopsy was not performed.
Discussion
Schinzel Giedion syndrome (SGS) is a rare genetic disorder with characteristic facial features, skeletal abnormalities, and ureteral obstruction which may lead to hydronephrosis. Symptoms characteristic of Schinzel Giedion syndrome include excessive hair-growth (hypertrichosis), a flat midface (midface retraction), seizures, clubfeet, broad ribs, profound intellectual disability and short arms and legs. Schinzel Giedion syndrome is caused by a de novo mutation of the SETBP1 gene, thus is not inherited from the parents. It is a severe progressive syndrome and most affected individuals do not survive infancy. The SETBP1 gene is a cancer promoting gene, and affected children who survive past three years of age are at risk for different types of cancer [1,2].
Prior to the discovery of the SETBP1 genetic marker, prenatal diagnosis relied on ultrasound examination, especially detection of renal abnormalities [3]. Hydronephrosis is detectable on prenatal ultrasound from week 18 to week 37, as in our case. A more recent case reported two Japanese patients with Schinzel–Giedion syndrome. When polyhydramnios is observed, additional fetal findings such as overlapping fingers, hydrocephalus, hydronephrosis, and very characteristic facial appearance comprising high, prominent forehead, hypertelorism, and depressed nasal root may suggest Schinzel–Giedion syndrome [4].
Hydronephrosis and various urogenital anomalies are common and may be important in establishing a clinical diagnosis. Touge et al reviewed 35 reported cases and observed hydronephrosis in 31 of them [5]. Hydronephrosis may also be detected during the antenatal period. Other urinary tract abnormalities include hydroureter, double ureter, pyeloureteral junction stenosis, ureterovesical junction stenosis, vesicoureteral reflux, and megacalyces. Congenital megacalycosis has been reported in four patients to date, one of whom was diagnosed prenatally. Congenital megacalycosis is believed to be to an abnormal development of the renal medulla leading to hypoplastic renal pyramids and blunted calyces. Megacalycosis may be unilateral or bilateral and is a nonprogressive dilatation of the calyces occasionally accompanied by lithiasis, infection, and hematuria, but with normal renal function. In most cases, the renal pelvis and ureters are not dilated, although primary megaureter can occur with megacalyces [6].
Reported nervous system findings comprise a wide spectrum of inconsistent abnormalities, including mild/moderate hydrocephalus, hypoplasia or agenesis of the corpus callosum, cerebral atrophy, absence of cranial nerves with normal cranial nuclei, and abnormal gyration of the cerebral cortex. Patients with Schinzel Giedion syndrome usually have trouble swallowing and breathing. Affected individuals do not survive after childhood, and the most common cause of death is pneumonia. Congenital cardiac defects, tumors, lung hypoplasia, intractable seizures, and sudden cardiac arrest are other causes of death seen during infancy [6].
References
[1] Schinzel A. "Schinzel Giedion Syndrome." National Organization for Rare Disorders (NORD). https://rarediseases.org/rare-diseases/schinzel-giedion-syndrome, Publish date 2017.
[2] Summerfield N. "How Schinzel-Giedion Syndrome (SGS) has changed our lives." Congenica. https://blog.congenica.com/how-schinzel-giedion-syndrome-has-changed-our-lives.
[3] Labrune P, Lyonnet S, Zupan V, et al. Three new cases of the Schinzel-Giedion syndrome and review of the literature. Am J Med Genet. 1994 Mar 1;50(1):90-3.
[4] Hishimura N, Watari M, Ohata H, et al. Genetic and prenatal findings in two Japanese patients with Schinzel–Giedion syndrome. Clin Case Rep. 2017 Jan; 5(1): 5–8.
[5] Touge H, Fujinaga T, Okuda M, et al. Schinzel-Giedion syndrome. Int J Urol. 2001 May;8(5):237-41.
[6] Bulut O, Ince Z, Altunoglu U, et al. Schinzel-Giedion Syndrome with Congenital Megacalycosis in a Turkish Patient: Report of SETBP1 Mutation and Literature Review of the Clinical Features. Case Rep Genet. 2017;2017:3740524.
Discussion Board
Winners

Anita Silber Israel Physician

Tudor Iacovache Romania Physician