Case of the Week #600
(1) Australia, Melbourne, Monash Ultrasound For Women; (2) Pediatric & Fetal cardiologist
A 37-year-old G2P1 woman presents with no family history of structural congenital heart disease. Both pregnancies were in vitro fertilization, with this pregnancy a result of frozen embryo transfer. The first child had an epiglottic cyst and spent two weeks in the intensive care unit after birth. The following images were obtained at 12 weeks, 4 days. Noninvasive prenatal testing was low risk and nuchal translucency was 1.5 mm.
Repeat ultrasound exam was obtained approximately 1 week later.
View the Answer Hide the Answer
Answer
We present a case of D-transposition of the great arteries with ventricular septal defect diagnosed in early pregnancy.
Our prenatal images identified:
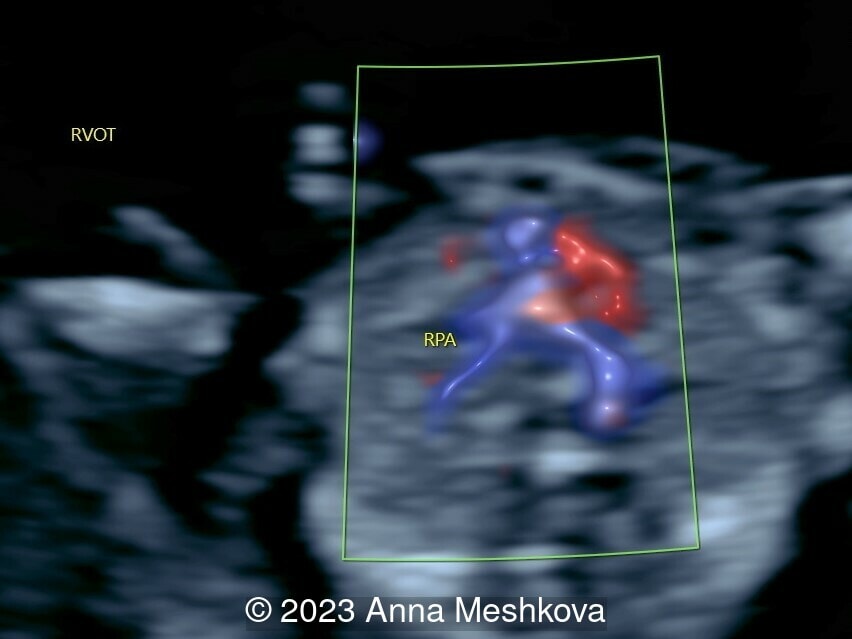
- The pulmonary artery with bifurcation into the left and right branches (Image 1)
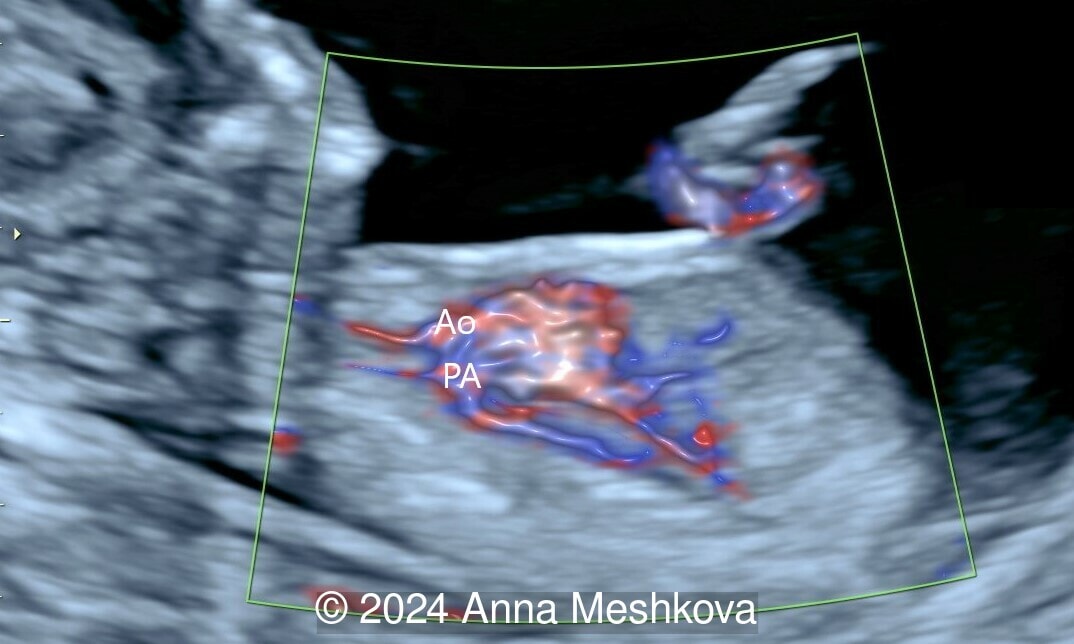
- 3 vessel view demonstrating that the aorta arises from the right ventricle (Video 1,2)
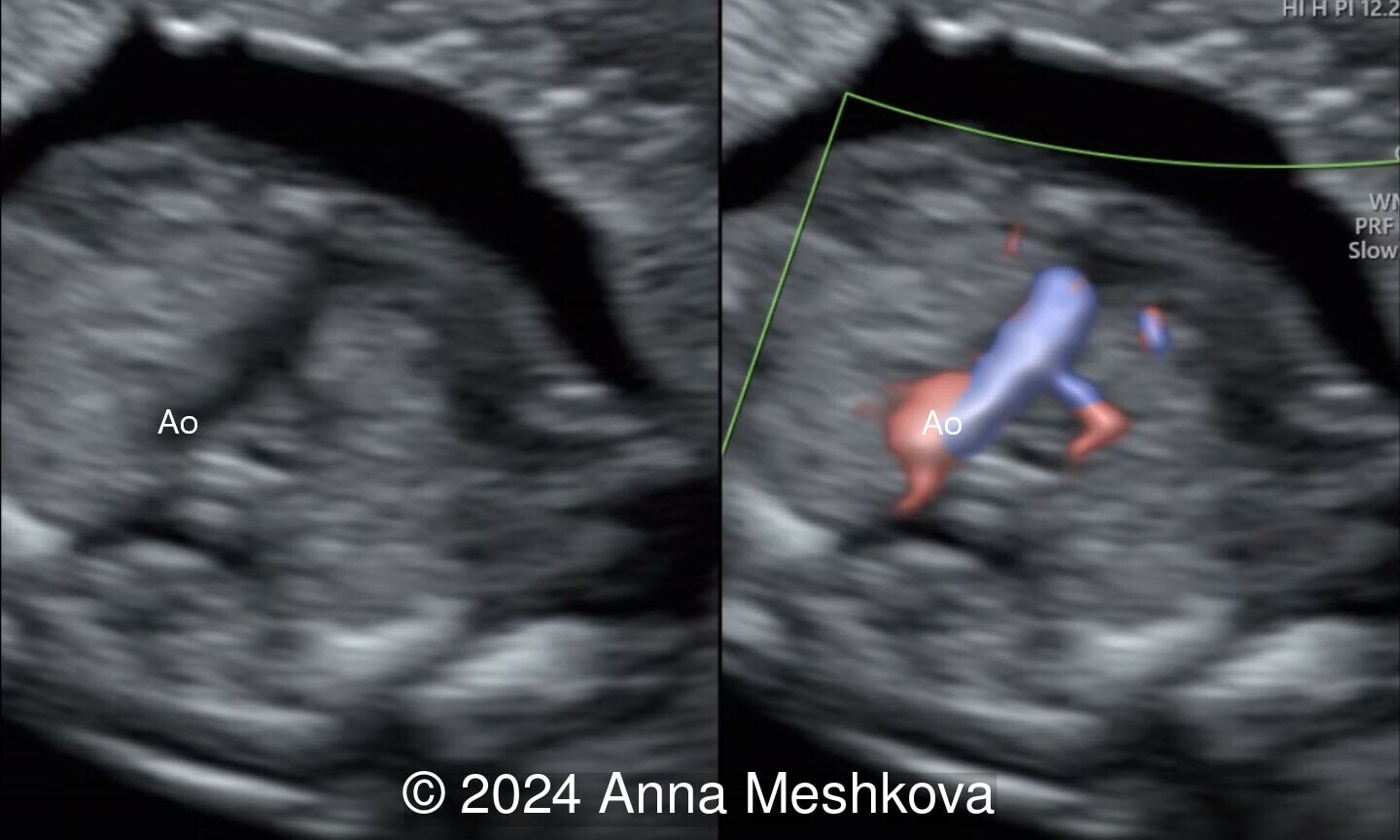
- Balanced ventricles with unobstructed outflow tracts and aorta arising from the right ventricle. Normal caliber of the left-sided aortic arch (Video 3).
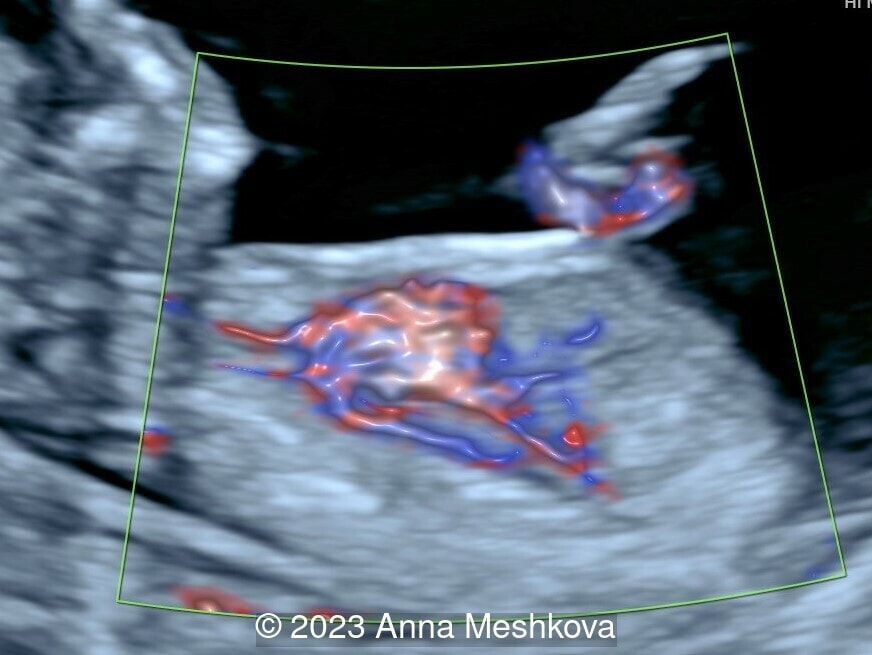
- Sagittal plane through aortic arch shows parallel course of the great vessels (Image 2, Video 6,7)
- High muscular perimembranous ventricular septal defect (video 4, 5)
- Normal ductus arteriosus with antegrade flow and confluent branch pulmonary arteries
- Mitral and tricuspid valvular function was normal with no atrioventricular valve regurgitation
- There was normal looking foramen ovale with right-to-left shunt
- There was normal systematic and pulmonary venous connections
- No markers of aneuploidy or extra-cardiac malformation was seen.
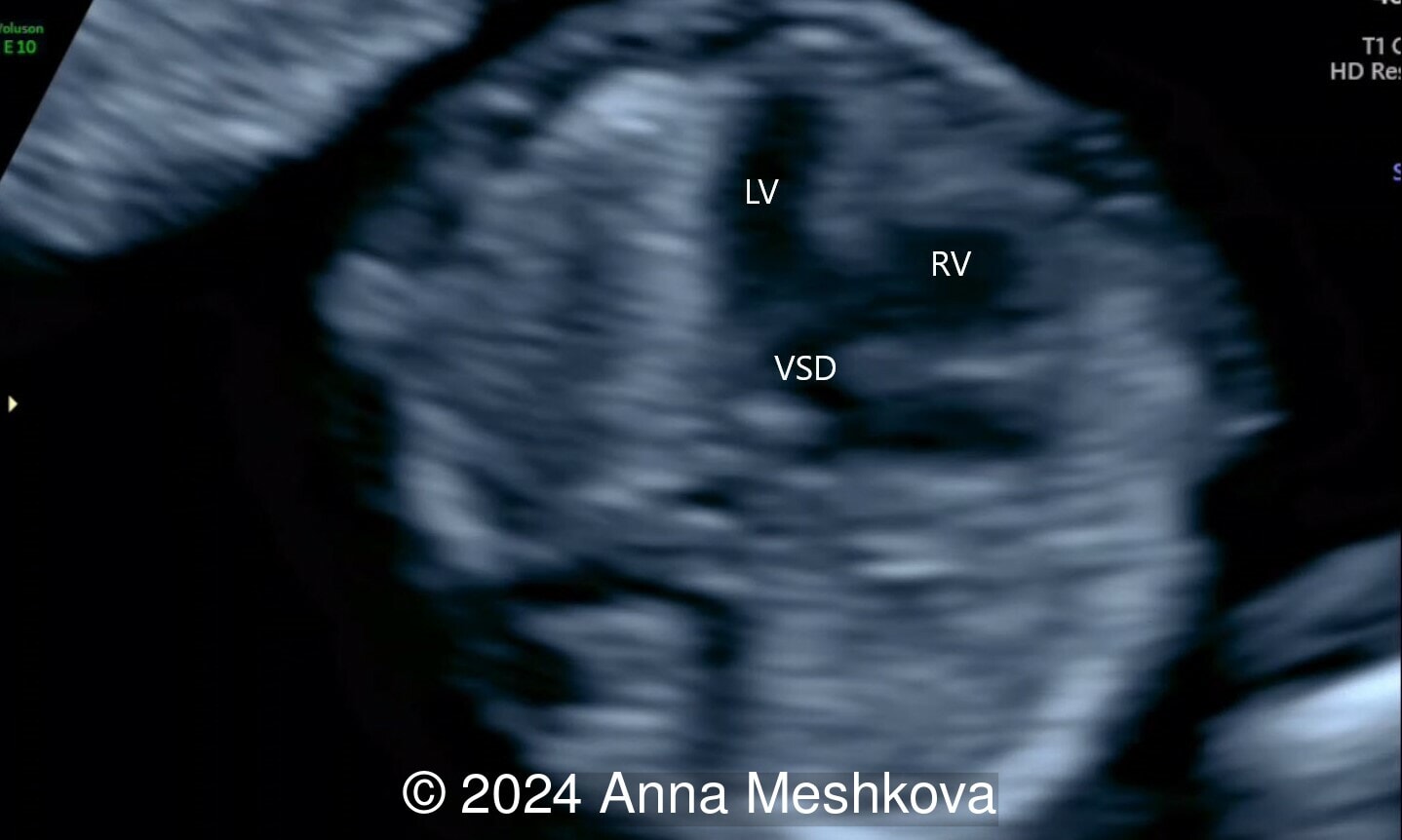
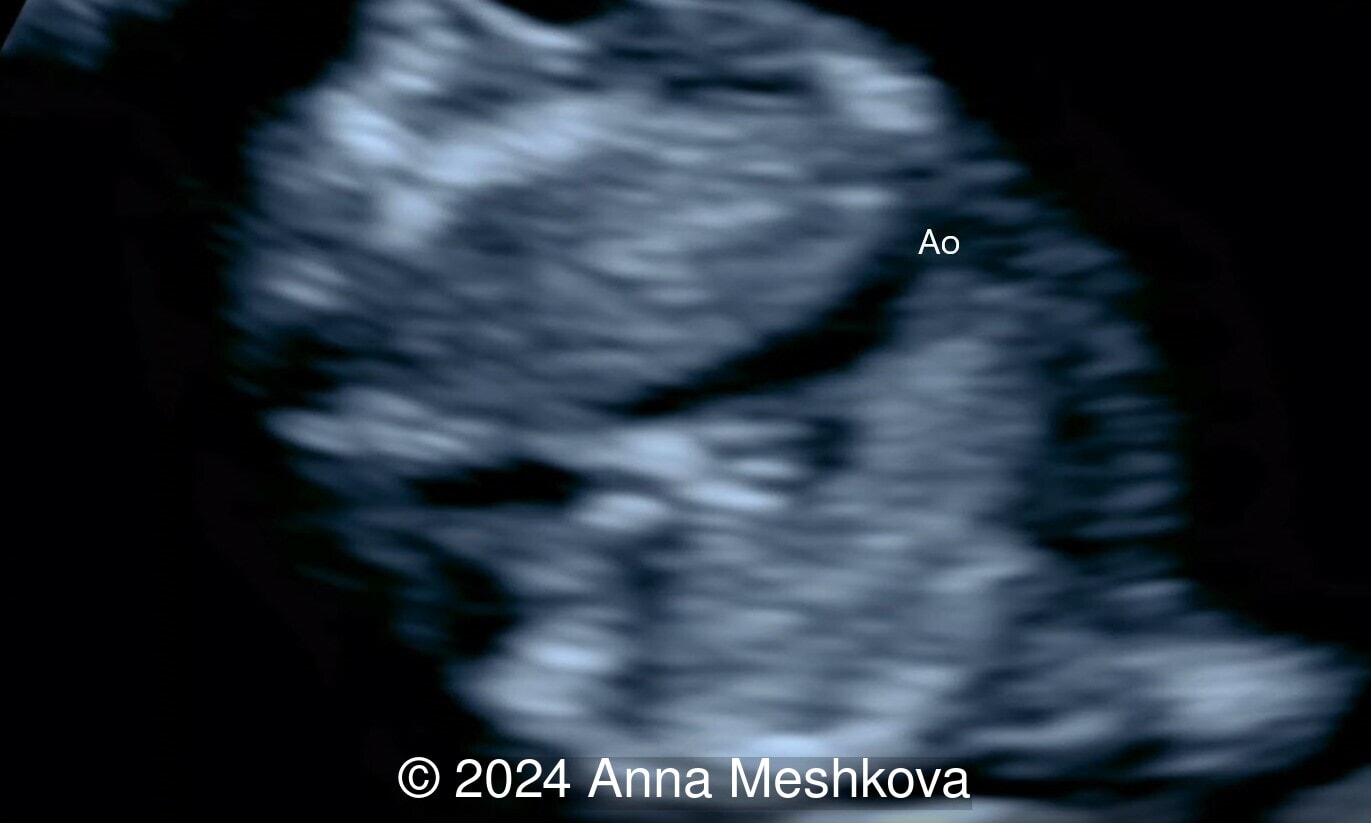
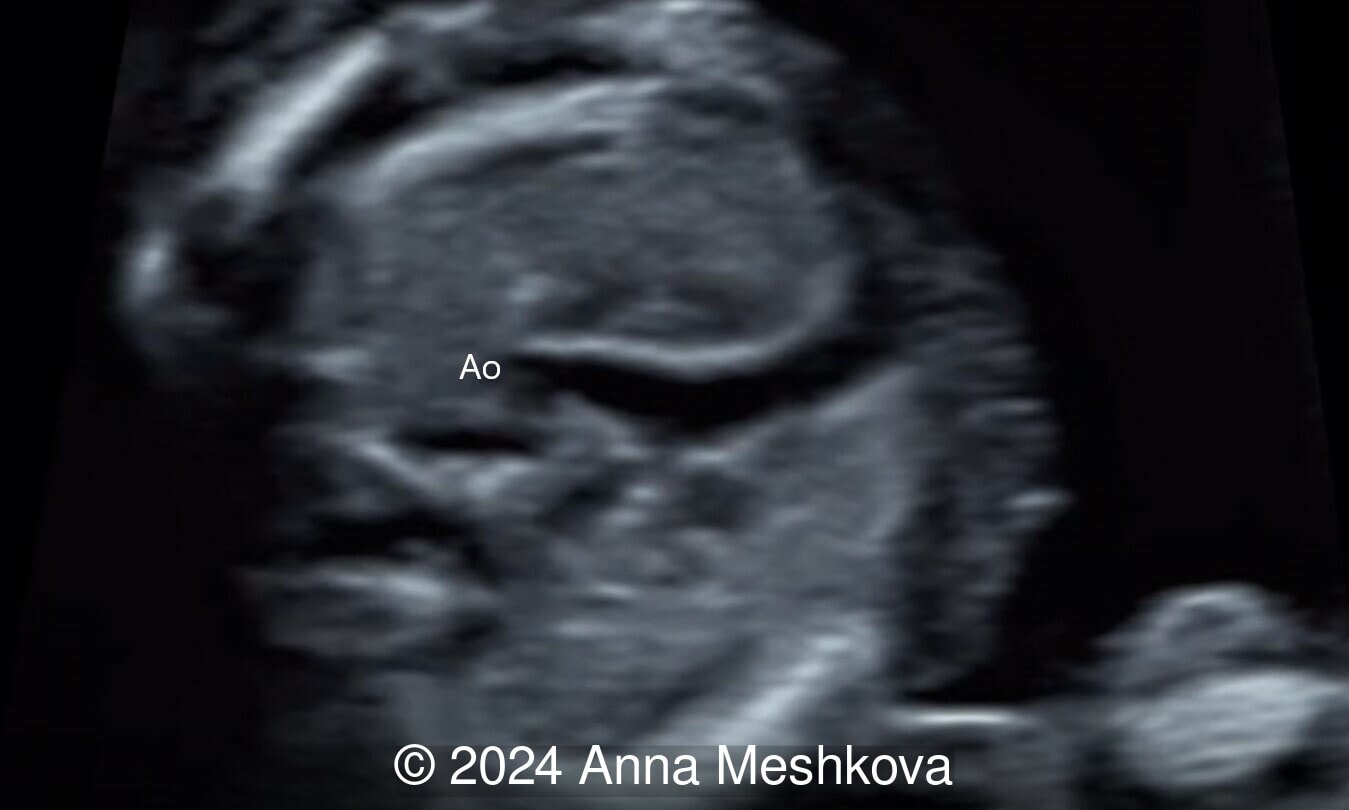
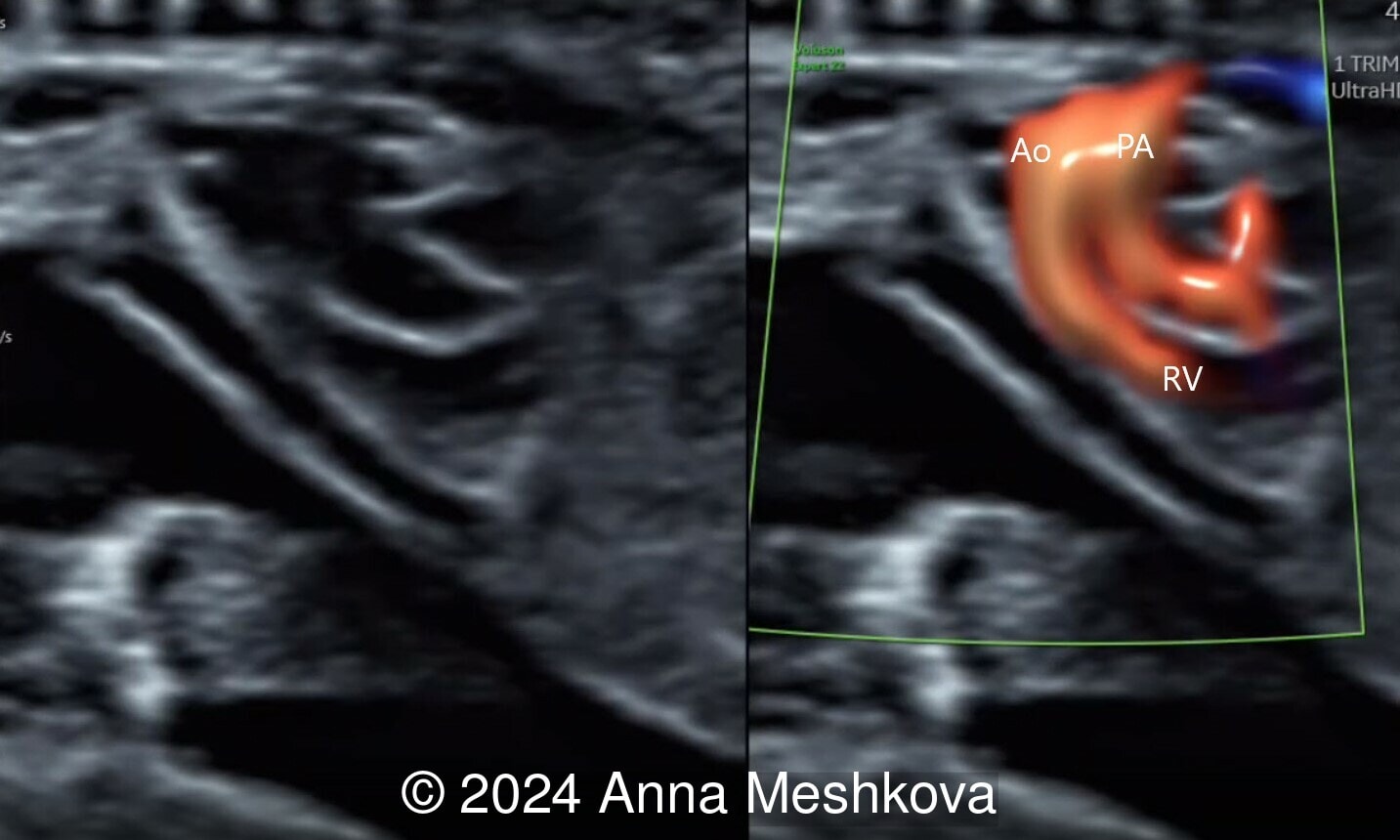
Discussion
Transposition of the great arteries is a common cardiac malformation with atrioventricular concordance (normal connection between atria and ventricles) and ventriculoarterial discordance (connection of the great vessels is switched). In our case, both great arteries display a parallel course, with the aorta anterior and to the right of the pulmonary artery (hence the term D-TGA “dexter”). D-transposition of the great arteries (D-TGA) is relatively frequent cardiac anomaly occurring in 5 to 7% of congenital cardiac malformations, with an incidence 0.315 cases per 1000 live births and 2:1 male preponderance.
Ventricular septal defect and pulmonary stenosis are common associations with D-TGA and may be present either alone or in combination in up to 30-40% cases. Associated extracardiac malformations as well as chromosomal abnormalities are rare with simple transposition of the great arteries.
On prenatal ultrasound, the four chamber view is typically normal, except for an associated ventricular septal defect. The aorta is noted to arise from the right ventricle in an anterior and parallel course to the pulmonary artery. Instead of a 3-vessel trachea view, a single large vessel (the transverse aortic arch) with a superior vena cava to its right is identified (I–sign). On longitudinal views, the aortic arch is seen arising from the anterior right ventricle, giving rise to head and neck vessels and assuming a “hockey stick” orientation as it curves posteriorly. The pulmonary artery assumes the “candy cane” orientation. Before inclusion of the outflow tracts to international fetal cardiac screening guidelines, the detection rates reported only 3 to 17 % of simple transposition of the great arteries. In one series of more than 3000 fetuses undergoing detailed screening for anomalies at 11 to 14 weeks, D-TGA is rarely diagnosed early in gestation [2]. The 3-vessel trachea view with a single great vessel may be helpful in early gestation. Additionally, an enlarged nuchal translucency in the setting of normal fetal chromosomes as well as an abnormal cardiac axis can be associated with conotruncal anomalies, including TGA.
Usually, babies with transposition of the great arteries with ventricular septal defect need to be to started on prostaglandin E1 infusion to maintain patency of the ductus arteriosus after birth. Some babies may require urgent balloon atrial septostomy to enlarge the foramen ovale if postnatal mixing is inadequate. Delivery at a tertiary center where neonatal intensive care facilities are available and capable of performing urgent septostomy is preferred. Surgical repair with an arterial switch and coronary translocation is normally performed at 1 week of age. If the ventricular septal defect allows good mixing within the heart, surgical correction can sometimes be deferred for 4-6 weeks, allowing babies to establish normal feeding and growth. The reported surgical mortality is approximately 1%. There is a higher incidence of learning difficulties and behavioral problems in children with transposition of the great vessels. The risks are greatest for babies born prematurely or with other associated extracardiac abnormalities. In the long term, a small number of patients require further surgery for the branch pulmonary arteries, which are placed over the top of the aorta as a part of surgery and are sometimes narrowed. Adult patients may require valvular repair or replacement due to neoaortic valve regurgitation as the original pulmonary root dilatates over many years.
Transposition of the great arteries can be a difficult diagnosis, but early identification allows more time for patient counselling, as well as planning pregnancy and delivery options.
References
1. Abuhamad A, Chaoui R. A practical guide to fetal echocardiography: normal and abnormal hearts, 3rd edition. Lippincott Williams & Wilkins, 2015. p447-466.
2. Becker R, Wegner RD. Detailed screening for fetal anomalies and cardiac defects at the 11–13-week scan. Ultrasound Obstet Gynecol. 2006 Jun;27(6):613-8.
Discussion Board
Winners

Guest United States

Dr. Hitanshu Bhatt India Physician

Dianna Heidinger United States Sonographer
Yulia Sologub Russian Federation Physician

Javier Cortejoso Spain Physician
Pawel Swietlicki Poland Physician
Umber Agarwal United Kingdom Maternal Fetal Medicine

Igor Yarchuk United States Sonographer
Larysa Gazarova United States Physician
belen garrido Spain Physician

Andrii Averianov Ukraine Physician

Ana Ferrero Spain Physician

Alexandr Krasnov Ukraine Physician

Mayank Chowdhury India Physician

Vladimir Lemaire United States Physician

Shilpen Gondalia India Physician

Boujemaa Oueslati Tunisia Physician

Tatiana Koipish Belarus Physician

CHARLES SARGOUNAME India Physician

Aysegul Ozel Turkey Physician

Philippe Deblieck Germany Physician

Kimberly Delaney United States Sonographer
Marianovella Narcisi Italy Physician
Javier Ayala Spain Physician
Rebecca Evans Australia Sonographer
Victoria Giang Viet Nam Physician
Muradiye YILDIRIM Turkey Physician

Ta Son Vo Viet Nam Physician
PATRICIA MUÑOZ United States Physician

ALBANA CEREKJA Italy Physician

Akmaral Medetbekova Kazakhstan Physician

Eti Zetounie Israel Sonographer

Murat Cagan Turkey Physician

Sonio Sonio France AI

Charlotte Conturie United States Physician
gholamreza azizi Iran, Islamic Republic of Physician

Ionut Valcea Romania Physician
aastha mehra India Physician

Viralkumar Madhu India Sonographer
Martina Vagaská Slovakia Physician

Anette Beverdam Netherlands Sonographer
Grzegorz Rak Poland Physician

Annette Reuss Germany Physician
Jana Furlan New Zealand Sonographer

shruti Agarwal India Physician

Mai Phương Viet Nam Physician

shay kevorkian Israel Physician
SAVITA SHIRODKAR India Physician
Nawar Hatoum United States Physician

Andrea Stoop-Berends Netherlands Sonographer

Ismail Guzelmansur Turkey Physician
Fatemeh Shakki Katouli Iran, Islamic Republic of Physician
Luciana Gimenez Spain Physician

Amy Scheible United States Sonographer
Flora Mirzoeva United States Sonographer
Rupal Sasani India Physician
Lashell Jones United States Sonographer
SHILPA KISHORE India Physician

Eylem Eşsizoğlu Turkey Physician
Süreyya Sarıdaş Demir Turkey Physician
Petra Barboríková Slovakia Physician

Carmen de Luis Rodríguez Spain Physician
Rajnikant Vasava India Physician

Denys Saitarly Israel Physician

Le Tien Dung Viet Nam Physician

Tetiana Ishchenko Ukraine Physician
Juliana Moren Argentina Physician
Jose Pablo Ling Garcia Mexico Physician
Vipalee Trivedi India Consult gynecologist
Gaurav Sharma India Physician
María Victoria Peral Parrado Spain Physician
Eduardo Namura Di Thomaz Brazil Physician

Le Duc Viet Nam Physician
Huỳnh Trí Trương Viet Nam Physician
Paul Timmons United Kingdom Physician
Erika Zanzarelli Italy Physician